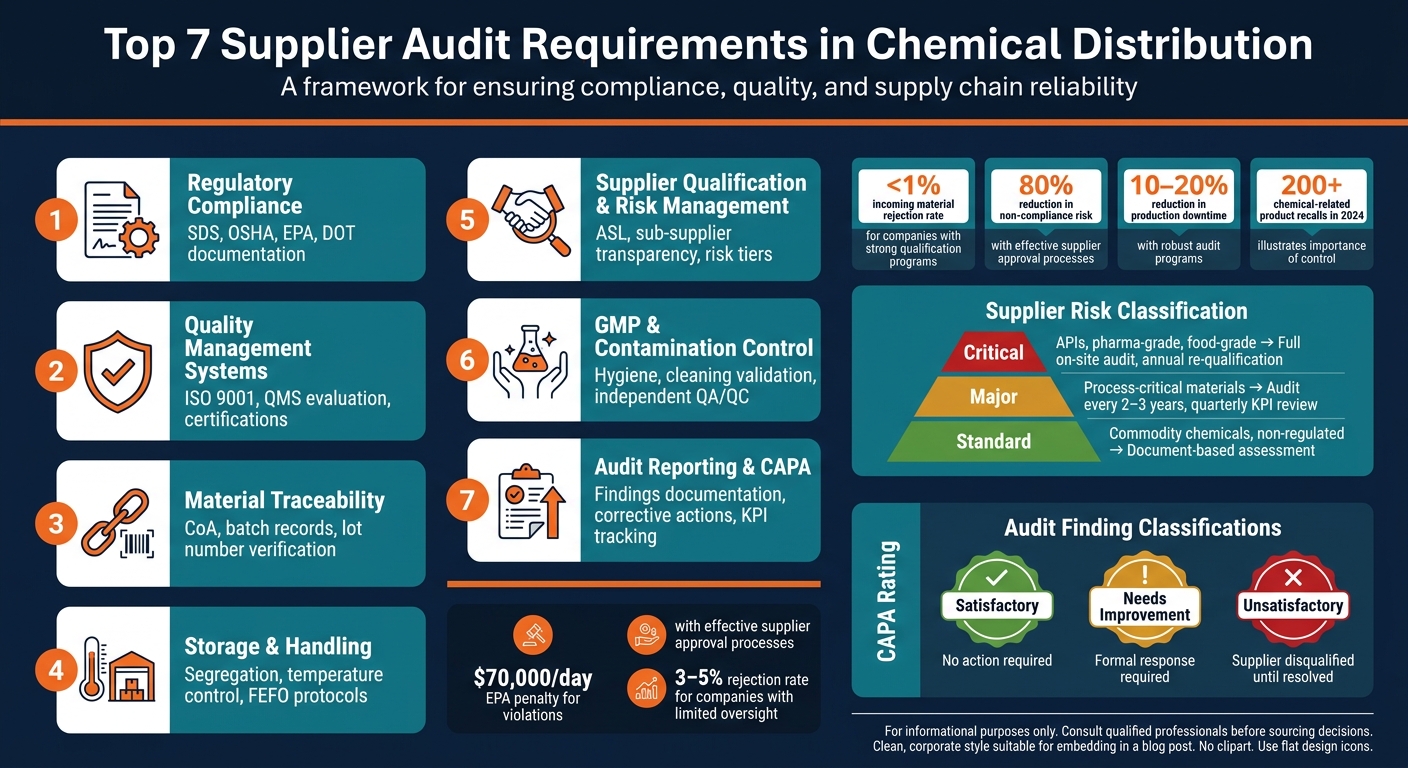
Auditing suppliers in chemical distribution is crucial for ensuring compliance, quality, and safety in highly regulated industries like pharmaceuticals, food, and electronics. A robust supplier audit process minimizes risks such as production delays, recalls, and regulatory penalties. Here are the seven key areas every supplier audit should cover:
- Regulatory Compliance: Suppliers must provide verified documentation like Safety Data Sheets (SDS) and meet OSHA, EPA, and DOT standards.
- Quality Management Systems: Evaluate internal systems, ensuring consistent processes and valid certifications specific to the materials being sourced.
- Material Traceability: Confirm documentation such as Certificates of Analysis (CoA) and batch records to ensure traceability and product quality.
- Storage and Handling: Inspect facilities for proper chemical segregation, temperature control, and adherence to safety protocols.
- Supplier Qualification and Risk Management: Maintain an Approved Supplier List (ASL), verify sub-supplier transparency, and classify vendors by risk level.
- GMP and Contamination Control: Ensure hygiene standards, validated cleaning protocols, and independent QA/QC oversight for sensitive applications.
- Audit Reporting and CAPA: Document findings, implement corrective actions, and continuously monitor supplier performance.
These steps help prevent disruptions, ensure product integrity, and build reliable supply chains. Knowing the right questions to ask chemical suppliers before signing a contract further strengthens these partnerships. Focusing on these areas reduces incoming material rejection rates and protects against costly compliance issues.
Supplier Audit: Strong vs. Weak Practices in Chemical Distribution
A Smarter Approach to Supplier Quality Audits with Thermo Fisher‘s David Festa
sbb-itb-aa4586a
1. Documented Regulatory Compliance Framework
Supplier audits in chemical distribution often begin with a critical question: Does the supplier have a clear, documented framework to prove regulatory compliance? Auditors need tangible evidence, as this framework is essential for meeting strict U.S. regulations.
In the United States, suppliers must navigate requirements from several regulatory bodies. For instance, OSHA’s Hazard Communication Standard (29 CFR 1910.1200) mandates that suppliers provide Safety Data Sheets (SDS) with all 16 sections fully completed. If a section doesn’t apply, it must explicitly state "no applicable information." Leaving fields blank raises immediate concerns during an audit [7].
For facilities handling highly hazardous chemicals, OSHA’s Process Safety Management (29 CFR 1910.119) requires detailed documentation, including written operating procedures, process hazard analyses (PHA), and a management of change (MOC) system. These requirements are particularly relevant for operations involving large quantities of flammable liquids (flashpoint below 100°F) [6].
Compliance doesn’t stop with OSHA. DOT regulations, outlined in SDS Section 14, require accurate transport classifications such as UN numbers, proper shipping names, hazard classes, and packing groups. Additionally, EPA and FDA regulations, where applicable, are addressed in SDS Section 15 [7].
"Assuming your chemical supplier is compliant is like betting your company’s future on a coin toss." – Kilburn Chemicals [8]
Maintaining current documentation is equally important. OSHA mandates revalidation of process hazard analyses at least every five years. Serious incident investigation reports must also be retained for five years, while operating procedures should be reviewed and certified annually to account for changes in equipment or processes [6]. A key question for auditors to ask is: "When was this last reviewed, and who conducted the review?"
This meticulous documentation forms the foundation of any effective audit framework.
| Regulatory Standard | Governing Body | Key Documentation |
|---|---|---|
| Hazard Communication (29 CFR 1910.1200) | OSHA | 16-section SDS, hazard labeling |
| Process Safety Management (29 CFR 1910.119) | OSHA | PHA, MOC procedures, incident reports |
| Transport Classification | DOT | UN number, shipping name, packing group (SDS Section 14) |
| Environmental & Health Regulations | EPA / FDA | Product-specific declarations (SDS Section 15) |
| Market Registration | EPA (TSCA) | TSCA inventory status verification |
2. Quality Management System and ISO Certifications
Once regulatory documentation is in place, the next step is evaluating a supplier’s internal quality systems as part of a chemical sourcing strategy. This involves assessing the supplier’s Quality Management System (QMS), which ensures consistent product quality in every delivery.
ISO 9001 certification is often considered the baseline for quality assurance in chemical distribution. Kilburn Chemicals explains:
"ISO 9001 certification confirms a documented, process-driven system for planning, controlling, and continuously improving operations." [4]
Suppliers with inadequate qualifications can experience significant issues. For example, 3–8% of API and intermediate orders from such suppliers fail testing, leading to production delays of 4–10 weeks and consuming 20–60 hours of QA resources per incident [5]. In contrast, organizations with well-developed supplier qualification programs report incoming material rejection rates of less than 1%, compared to 3–5% for those with limited oversight [2].
Auditors play a key role in verifying the scope of ISO 9001 certifications. They must ensure the certification applies to the specific site and chemical grade being sourced [1]. Additionally, it’s essential to confirm the issuing certification body, check the certificate’s validity date, and ensure that Certificates of Analysis (CoA) are signed by a qualified quality control professional rather than administrative staff [1].
While ISO 9001 provides a strong foundation for documentation in regulated industries, it does not replace the need for Good Manufacturing Practice (GMP) when dealing with pharmaceutical-grade materials. As ChemContract highlights, "ISO 9001 is necessary but not sufficient for any supplier producing pharmaceutical-grade material." [5]
For example, Allan Chemical Corporation operates under both ISO 9001 and ISO 14000 systems, showcasing a commitment not only to quality but also to environmental responsibility. This is particularly important given that Environmental Protection Agency (EPA) penalties can reach $70,000 per day for violations [3].
| Standard | Primary Focus | What It Confirms |
|---|---|---|
| ISO 9001 | General quality management | Consistent processes, continuous improvement, operational discipline |
| ISO 14001 | Environmental management | Reduced environmental liability, ESG compliance |
| ISO 17025 | Laboratory competence | Analytical testing and calibration accuracy |
| GMP | Manufacturing/testing environment control | Product safety and purity for regulated industries |
3. Material Traceability, Documentation, and Certificates
Auditors carefully examine the documentation that ensures material traceability after confirming a supplier’s quality management system. This step is crucial because it ties each product shipment to its quality data, offering proof that the material meets required specifications and FDA GMP standards.
A key document in this process is the Certificate of Analysis (CoA). This certificate provides batch-specific details such as numerical test results, analytical methods (like HPLC or GC), lot numbers, and manufacture and expiration dates [1][8]. A verified CoA replaces guesswork with hard data, minimizing the risk of regulatory issues or recalls. It also supports cross-referencing and maintains the chain of custody as outlined in 21 CFR 211.84.
The lot number plays a vital role – it acts as a unique identifier for each batch. Auditors must ensure that the lot number matches across the CoA, packaging, and shipping manifest, confirming the product’s traceability.
In addition to the CoA, a comprehensive documentation package is required. For example, the Safety Data Sheet (SDS) must adhere to GHS Revision 7 or later, as outdated versions can lead to customs delays or regulatory complications. The Certificate of Compliance (CoC) verifies that the material meets regulatory standards like TSCA or EU REACH, while batch records provide a detailed manufacturing history, including the equipment used, process steps, operator signatures, and timestamps. Together, these documents ensure a robust traceability system that meets regulatory requirements.
Suppliers like Allan Chemical Corporation include CoAs and SDSs as standard documentation, helping customers in regulated industries streamline their material reviews. However, certain red flags can signal potential issues. For instance, a CoA signed by administrative staff rather than a qualified quality control professional raises concerns. Auditors should also insist on obtaining documents directly from the manufacturer to avoid altered or fraudulent records. Additionally, an internal batch rejection rate higher than 5% suggests quality issues, as reputable manufacturers typically report rates between 1% and 3%.
These documentation practices are essential for maintaining supplier integrity and ensuring regulatory compliance.
| Document | Purpose | Key Elements to Verify |
|---|---|---|
| Certificate of Analysis (CoA) | Confirms batch-specific quality and purity | Lot number, test methods, results, quality signature |
| Safety Data Sheet (SDS) | Details hazards and handling requirements | GHS Revision 7+ compliance, chemical identity |
| Certificate of Compliance (CoC) | Verifies regulatory compliance | TSCA/REACH status, manufacturer details |
| Batch Records | Tracks full manufacturing history | Equipment, process steps, operator signatures, timestamps |
| Certificate of Origin (CoO) | Identifies country of manufacture | Issued by a chamber of commerce, not self-declared |
4. Chemical Safety, Handling, and Storage Controls
After verifying documentation, auditors turn their attention to the physical spaces where chemicals are stored and handled. Even the best paperwork cannot offset poor storage practices that might jeopardize material quality or safety. This transition from reviewing records to inspecting physical conditions is a key step in maintaining compliance.
Facility inspections are a major focus here. Auditors evaluate warehouses for cleanliness, proper organization, and pest control measures. They also confirm that environmental monitoring equipment is correctly calibrated. To ensure compliance, they review a full year’s worth of temperature and humidity logs against specified thresholds.
Maintaining proper material segregation is another critical aspect. Materials must be separated by their status – whether they are received, quarantined, released, returned, or rejected. Clear systems like color-coded markings, signage, and physical barriers are essential. Additionally, software systems should enforce restrictions on quarantined stock to prevent accidental use.
"GMP storage is the discipline of holding regulated materials and finished products under conditions that preserve their identity, strength, quality, and purity from the moment they arrive at a warehouse until they ship to the next stage of the supply chain." – Kurt Adams, Author [10]
Temperature and humidity control are closely examined during audits. For instance, pharmaceutical-grade chemicals typically require humidity levels below 65% relative humidity to prevent degradation of moisture-sensitive (hygroscopic) materials [10]. Auditors also expect temperature mapping studies conducted with calibrated data loggers to identify any hot or cold spots in storage areas. For chemicals that require temperature control, facilities must have backup refrigeration and power systems in place.
Hazardous material handling and chemical safety go through a separate layer of scrutiny. Auditors check that SDS (Safety Data Sheets) are easily accessible and confirm that personnel managing hazardous substances – like corrosives, flammables, or oxidizers – follow proper procedures and wear the recommended PPE. For packaged hazardous materials, UN test certificates must be available to verify compliance. Additionally, facilities must demonstrate that they follow FEFO (First-Expiry, First-Out) inventory protocols, ensuring older stock is used or shipped before newer inventory.
Strong storage practices work hand-in-hand with documented processes, creating a comprehensive approach to compliance. At Allan Chemical Corporation, these strict measures help maintain product integrity throughout storage, reinforcing the quality standards upheld at every step of the supply chain.
5. Supplier Qualification, Sub-supplier Control, and Risk Management
Once physical storage and handling practices are verified, auditors focus on how suppliers and sub-suppliers are managed. The strength of a chemical distributor’s quality often depends on its supply chain, which can extend through multiple tiers.
Auditors expect to see a formal Approved Supplier List (ASL) that is actively maintained and updated with performance data. Suppliers who fail to meet quality standards or consistently miss delivery deadlines should be flagged for probation or removal. This is particularly critical when working with pharmaceutical-grade or food-grade chemicals, where precision and reliability are non-negotiable.
Sub-supplier transparency is another major focus. Distributors must confirm the true origins of raw materials instead of relying solely on Certificates of Analysis (CoAs) provided by manufacturers. This step strengthens traceability efforts by ensuring accurate origin information.
Change control protocols are also reviewed to ensure that any changes to manufacturing processes, formulations, or packaging trigger a mandatory 90-day notification. This precaution helps avoid downstream issues like batch failures or regulatory non-compliance [4].
Lastly, auditors examine how vendors are classified based on risk. For example, critical materials like active pharmaceutical ingredients (APIs) require stricter oversight compared to standard packaging materials. Suppliers operating in regions with weaker regulatory frameworks may need more frequent audits and additional incoming quality checks. At Allan Chemical Corporation, this structured approach to supplier management ensures the consistency and traceability that industries with strict regulations rely on, helping to mitigate risks across the supply chain.
6. GMP, Hygiene, and Contamination Control for Sensitive Applications
Auditors closely examine chemical handling processes in industries like pharmaceuticals, food production, and cosmetics, where even the smallest contamination can lead to serious issues. Similar to earlier supplier assessments, they ensure there is independent QA/QC oversight, which prevents any compromises driven by production demands. This independent team must have the authority to approve or reject materials, review batch records, and identify errors. Naturally, this scrutiny extends to the conditions of the facility and the practices of its employees.
Facilities need to implement cross-contamination prevention methods, such as advanced air filtration systems, while employees are required to wear protective clothing and complete GMP (Good Manufacturing Practice) training. As outlined in USP <1078>:
"Any person shown at any time… to have an apparent illness or open lesions that may adversely affect the safety or quality of excipients should be excluded from direct contact with components." [11]
For equipment used across different product grades, validated cleaning protocols are critical. Surfaces in contact with products must not react chemically, add contaminants, or retain residues. Cleaning logs must document prior batches to confirm proper cleaning, which is especially crucial for distributors like Allan Chemical Corporation. They supply compendial-grade chemicals – such as USP, FCC, and NF grades – where batch carryover can directly affect quality.
Here’s a summary of the key GMP focus areas typically evaluated in sensitive-application environments:
| GMP Focus Area | Key Requirement |
|---|---|
| Personnel | GMP and hygiene training; use of protective clothing like head and arm coverings |
| Facilities | Air systems to prevent cross-contamination; pest-free environment |
| Equipment | Validated cleaning procedures for shared lines; non-reactive contact surfaces |
| Traceability | Batch numbering for complete tracking; recall capabilities |
| Quality Control | Independent QA/QC team; retention of samples for 1 year after expiration |
| Sanitation | Written cleaning schedules; plumbing designed to prevent back-siphonage |
Beyond these core GMP areas, environmental monitoring plays a critical role in maintaining standards for sensitive applications. This includes controlling exposure to light, heat, and air. When inert gases are used during processing, they must be treated as raw materials and tracked with the same level of detail. Such measures ensure that the integrity and safety of products remain uncompromised.
7. Audit Reporting, CAPA, and Continuous Improvement
An audit should result in a clear and actionable report. According to 21 CFR § 117.475, receiving facilities are required to document specific details, including the supplier’s name, audit procedures, audit dates, conclusions, and any corrective actions taken [9]. A thorough audit report must also include the supplier’s legal name, site address, D-U-N-S number, risk classification (Low, Medium, or High), and administrative sign-offs from both Quality Assurance and Procurement management [2]. By building on findings from earlier evaluations, this reporting phase ensures accountability across the supply chain and lays the groundwork for corrective actions and measurable progress.
Each audit finding is categorized as Satisfactory, Needs Improvement, or Unsatisfactory. A "Satisfactory" rating requires no further action. "Needs Improvement" highlights significant issues that demand a formal response, while "Unsatisfactory" points to critical deficiencies that disqualify the supplier until those issues are resolved and verified. Final audit conclusions align with these ratings, resulting in the supplier being classified as Approved, Conditionally Approved (with remediation required within 30–90 days), or Not Approved [2].
The CAPA (Corrective and Preventive Action) process plays a pivotal role in addressing immediate concerns and preventing future occurrences [12]. A well-executed CAPA involves root cause analysis, corrective measures, verification of effectiveness, and formal closure [12]. Superficial explanations, like attributing issues to "human error", are insufficient:
"If a supplier’s root cause analysis says nothing more than ‘human error’ or ‘operator mistake,’ that’s a major warning sign." – ChemXpert [12]
To ensure credibility, CAPA must be backed by solid evidence, such as updated SOPs, change control records, or validation reports. As ChemXpert emphasizes:
"Documentation proves the CAPA was implemented, not just planned. If a supplier can’t produce supporting evidence, the CAPA exists only on paper." [12]
High-performing suppliers typically respond to quality issues or deviation notices within 10 days. Audit findings should feed directly into KPI scorecards that track metrics like On-Time In-Full (OTIF) delivery (target: >95%), Out-of-Specification (OOS) rates, and documentation completeness (target: 100%) [2][4]. Addressing issues through CAPA and continuously monitoring performance strengthens the supplier audit framework.
Re-qualification processes should not rely solely on scheduled dates. Instead, immediate re-audits should be triggered by significant events, such as changes in ownership, manufacturing site relocations, or two consecutive lot rejections [2]. Companies with well-developed audit and qualification programs often report incoming material rejection rates below 1%, compared to 3%–5% for those with less oversight [2].
Disclaimer: This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Comparison Table
To make supplier audits more effective, the table below highlights the differences between strong and weak practices across several key audit areas. A robust audit ensures thorough process verification, while weaker audits often rely solely on documentation. Use this as a reference to identify potential gaps in your supplier evaluations.
| Audit Area | Strong Practice | Weak Practice |
|---|---|---|
| CoA Generation | Auto-generated from validated LIMS; results independently verified [4] | Manual data entry into spreadsheets or notebooks; no independent testing [4] |
| Change Management | Mandatory 90-day advance notice for any process, formulation, or packaging changes [4] | No formal notification; changes discovered only upon delivery [4] |
| Traceability | Batch-level tracking verified through active mock recall drills; physical retention samples maintained for shelf-life duration [4] | Lot-level tracking only; no retain samples kept for dispute resolution [4] |
| Testing Methods | Specific methods cited (e.g., HPLC, Karl Fischer, GC); in-house lab with accredited third-party backup [1] | Generic "as per standard" descriptions; all testing outsourced with no internal verification [1] |
| Regulatory Status | REACH, GHS, and sanctions list status independently verified [1] | Reliance on supplier self-declarations without independent verification [1] |
| Logistics & Packaging | UN-certified packaging with verified test certificates [1] | Self-declaration of compliance without supporting documentation [1] |
| Commercial Terms | Includes penalty clauses for quality failures and delivery delays [1] | "Best efforts" language with no financial accountability [1] |
| Inspection Stance | Welcomes third-party pre-shipment inspections (PSI) by agencies such as SGS or Intertek [1] | Refuses or resists third-party origin inspections [1] |
For better resource allocation, suppliers can also be categorized by risk level. This helps prioritize audits based on the potential impact on product quality or safety.
| Risk Tier | Criteria | Audit Requirement |
|---|---|---|
| Critical | High impact on product safety, purity, or efficacy (e.g., active ingredients, food-grade, pharmaceutical-grade chemicals) | Full on-site audit; annual re-qualification; batch-level CoA verification [4] |
| Major | Significant impact on manufacturing process or product stability; multiple sources available | Periodic on-site or remote audit every 2–3 years; quarterly KPI scorecard review [4] |
| Standard | Low impact on final product; widely available commodity chemicals; non-regulated applications | Initial document-based assessment; periodic questionnaire updates [4] |
"A single off-spec container load can shut down a production line for days." – RawSource [1]
For new or critical suppliers, third-party pre-shipment inspections (PSI) by agencies like SGS or Intertek provide an extra layer of verification. These inspections cost between $300–$800 per shipment, a small price compared to the potential fallout from production delays or regulatory issues [1].
Disclaimer: This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Conclusion
Supplier audits play a crucial role in chemical distribution, addressing key areas like regulatory compliance, quality, traceability, and safety. Together, these elements create a framework that ensures supply chains remain reliable and products maintain their integrity.
Effective supplier approval processes can lower non-compliance risks by as much as 80% [3] and cut production downtime by 10–20% [3]. With over 200 chemical-related product recalls reported in 2024 alone [3], the importance of robust supplier qualification cannot be overstated.
"Supplier qualification done well is unglamorous, repeatable, and quietly compounding – the firms with the best qualification programs spend less time fighting fires, win more inspections, and consolidate spend with fewer better-qualified suppliers." – Dr. Ram Sharma, Founder & CEO, ChemContract [5]
Dr. Sharma’s words highlight the value of disciplined audit programs. For distributors operating in regulated sectors like pharmaceuticals, food, cosmetics, and electronics, this level of thoroughness is non-negotiable. Allan Chemical Corporation supports its customers by providing access to thoroughly vetted manufacturers and detailed documentation, including Certificates of Analysis and Safety Data Sheets, across both compendial and technical-grade product offerings.
A strong audit program not only ensures product quality but also protects production schedules, regulatory compliance, and customer confidence.
Disclaimer: This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
FAQs
How often should we re-audit a chemical supplier?
Re-audit schedules should align with a risk assessment considering the material’s importance and the supplier’s criticality. Federal regulations mandate annual on-site audits for high-risk materials. For lower-risk items, the timing depends on factors like performance monitoring and the supplier’s risk profile. Allan Chemical Corporation emphasizes the value of continuous performance tracking and reviewing third-party audit reports to maintain quality and regulatory compliance.
What documents should we require with every shipment?
Each shipment must come with two essential documents: a Certificate of Analysis (COA), which provides batch-specific details like purity and composition, and a Safety Data Sheet (SDS), which explains handling, storage, and emergency procedures. For added traceability, you might also request a Technical Data Sheet (TDS) to review product specifications and a Certificate of Compliance (COC) to verify regulatory adherence.
When is an on-site supplier audit necessary?
An on-site supplier audit becomes essential when a supplier handles hazards that could threaten human or animal health, as outlined by FDA regulations such as PCHF, PCAF, and FSVP. These audits are especially important for high-risk materials or when working with new suppliers. They help confirm facility conditions, staff qualifications, and the robustness of quality systems. While reviewing documentation is important, on-site audits provide an unbiased and thorough evaluation that cannot be achieved through paperwork alone.



Comments are closed