Raw material testing is a critical step for ensuring the safety, quality, and compliance of pharmaceutical, dietary supplement, and food products in the U.S. The FDA’s Good Manufacturing Practice (GMP) regulations (21 CFR Parts 210, 211, and 111) require manufacturers to quarantine, test, and approve all raw materials – referred to as "components" – before use. Noncompliance can lead to serious consequences, such as recalls, fines, or criminal charges.
Here’s what you need to know:
- Identity Testing: Every raw material batch must undergo at least one specific test to verify its identity, even if a supplier provides a Certificate of Analysis (CoA). Sampling must follow statistical protocols, and documentation must be thorough and real-time.
- Microbiological and Purity Testing: Materials must meet strict limits for microbial contamination and impurities. High-risk components, like Glycerin and Propylene Glycol, now require mandatory testing for contaminants such as Diethylene Glycol (DEG) and Ethylene Glycol (EG).
- Supplier Qualification: Manufacturers must evaluate and approve suppliers, ensuring traceability back to the original source. CoAs can streamline testing but must be regularly verified through independent checks.
- Data Integrity: Laboratory systems must secure data with individual passwords and maintain audit trails to prevent tampering.
The FDA’s focus on quality control means manufacturers must maintain rigorous testing protocols, independent quality units, and detailed records to pass inspections and avoid regulatory actions.
Identity Verification Testing under 21 CFR 211.84
Identity Testing Requirements
According to 21 CFR 211.84(d), manufacturers are required to perform "at least one test…to verify the identity of each component of a drug product. Specific identity tests, if they exist, shall be used." This regulation applies to every batch of raw materials, containers, and closures that enters the facility. Even if a Certificate of Analysis is provided by a trusted supplier, manufacturers must still conduct at least one specific identity test on-site.
The Quality Control (QC) team plays a crucial role in this process. Materials cannot be used until they are sampled, tested, and officially released. Until these steps are completed, each lot is held in quarantine. Sampling must follow statistically defined protocols, taking into account factors such as component variability, confidence levels, and the supplier’s quality history. To prevent contamination during sampling, containers should be cleaned before opening, aseptic techniques applied when necessary, and resealed immediately afterward.
Samples from different levels within a container must be tested separately to account for variability. For drug product containers and closures, a visual inspection is considered the bare minimum, even when relying on a supplier’s certificate of testing. These identity verification steps are foundational for subsequent quality checks, including microbiological and purity assessments.
Documentation Requirements for Identity Verification
Identity testing must be documented at the time it is performed, as stated in 21 CFR 211.160(a): "The requirements in this subpart shall be followed and shall be documented at the time of performance. Any deviation from the written specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms shall be recorded and justified." This real-time documentation ensures an auditable trail for FDA inspections.
Each sample should be labeled with critical details such as the material name, lot number, sampling date, and the collector’s name. Original containers must also be marked for traceability. Test methods must be validated, and instruments calibrated according to a written program. If a supplier’s report of analysis is used for testing purity or strength, its reliability must be periodically confirmed through in-house testing. Proper documentation not only supports regulatory compliance but also helps streamline FDA audits.
This content is for informational purposes only. Always consult official regulations and qualified professionals before making sourcing or formulation decisions.
sbb-itb-aa4586a
Raw Material Qualification Process
Microbiological and Purity Testing Requirements
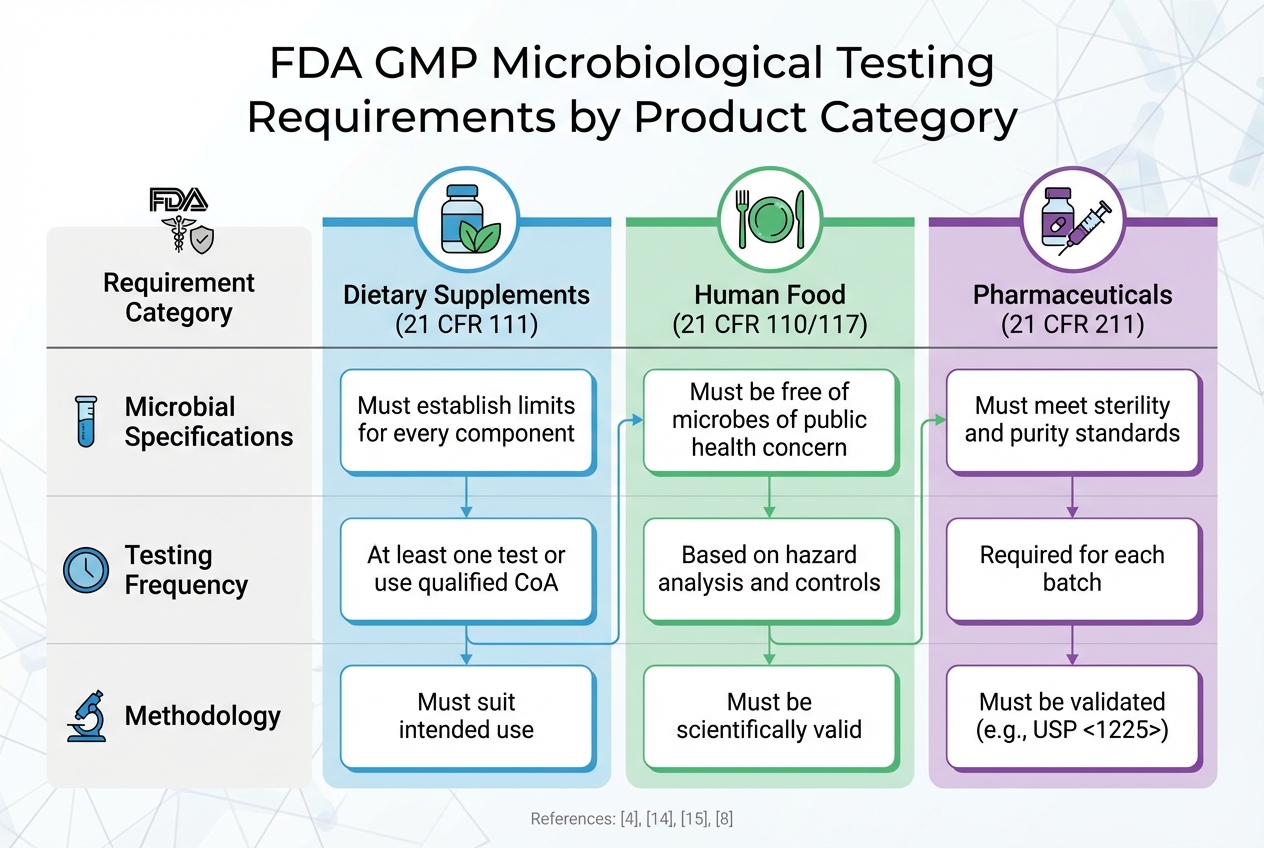
FDA GMP Microbiological Testing Requirements Comparison Table
Microbiological Testing Standards
After confirming raw material identity, microbiological and purity testing are critical steps in maintaining GMP compliance. These processes ensure that raw materials meet strict microorganism limits. According to 21 CFR Part 110.80, "Raw materials and other ingredients shall either be free of microorganisms of public health significance, or they shall be treated during manufacturing operations so that they no longer contain levels that risk product adulteration." [5] This regulation applies to pharmaceutical, food, and dietary supplement production.
The testing method depends on the product type and associated risks. For pharmaceuticals, sterility, endotoxin, and bioburden testing require validation [4]. Even if using pre-validated USP methods, manufacturers must confirm their effectiveness in their own facilities [4]. Bioburden testing is especially important for materials used in injectable or sterile drugs, as it assesses both endotoxin levels and sterility [4].
Environmental monitoring is another essential safeguard against contamination. Facilities must check for pathogens like Listeria monocytogenes and Salmonella in areas where raw materials are exposed, particularly when those materials are prone to contamination [6]. Personnel hygiene also plays a significant role. Under 21 CFR Part 111.10, "You must take measures to exclude from any operations any person who might be a source of microbial contamination, due to a health condition, where such contamination may occur, of any material, including components." [3]
| Requirement Category | Dietary Supplements (21 CFR 111) | Human Food (21 CFR 110/117) | Pharmaceuticals (21 CFR 211) |
|---|---|---|---|
| Microbial Specs | Must establish limits for every component [3] | Must be free of microbes of public health concern [5] | Must meet sterility and purity standards [4] |
| Testing Frequency | At least one test or use qualified CoA [3] | Based on hazard analysis and controls [6] | Required for each batch [4] |
| Methodology | Must suit intended use [3] | Must be scientifically valid [6] | Must be validated (e.g., USP <1225>) [4] |
These microbiological safeguards work in tandem with purity testing to ensure raw material safety.
Purity Testing for Contaminants and Impurities
Purity testing builds on microbiological standards by verifying that raw materials consistently meet quality specifications. This is a key requirement under CGMP regulations in 21 CFR Parts 210 and 211. Manufacturers must use validated analytical methods to detect impurities, as older methods may fail to identify modern contaminants.
In May 2023, the FDA issued guidance for testing high-risk drug components, such as Glycerin, Propylene Glycol, Maltitol Solution, and Sorbitol Solution, for toxic substances like Diethylene Glycol (DEG) and Ethylene Glycol (EG) [1]. This guidance aims to prevent fatal contamination events caused by these substances entering the supply chain. Manufacturers are required to test specifically for these contaminants rather than relying solely on supplier certificates.
If Out-of-Specification (OOS) results occur, they must be investigated and documented within 20 business days [4]. The FDA explicitly states that "Products cannot be ‘tested into compliance’ by arbitrarily labeling out-of-specification lab results as ‘laboratory errors’ without an investigation resulting in scientifically valid criteria." [4] Additionally, averaging OOS and passing results to achieve compliance is prohibited, as it conceals variability and violates FDA standards [4].
For sterile products, water quality is tightly controlled. Water used in the final isolation and purification of nonsterile APIs intended for sterile drugs must be tested for microbial counts, objectionable organisms, and endotoxins. Equipment that contacts raw materials must also be monitored to ensure it does not react with or compromise material purity.
This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Supplier Qualification and Certificate of Analysis Reliance
Supplier Qualification Process
Qualifying raw material suppliers is a key FDA GMP requirement, ensuring suppliers are thoroughly evaluated and approved before their materials are used. An independent quality unit must oversee and document procedures for receiving, identifying, sampling, and testing incoming materials. These records must include critical details like the component name, manufacturer, lot number, and receiving date to ensure traceability [2].
The FDA makes a clear distinction between the "supplier" (the entity providing the material) and the "manufacturer" (the entity producing it). Even if materials are purchased through third-party distributors or brokers, it’s essential to maintain traceability back to the original manufacturer [2]. This includes detailed records of all facilities involved in the supply chain. Beyond chemical testing, suppliers should also be assessed for critical physical attributes like particle size or moisture content, which can directly affect production consistency [2]. These rigorous qualification processes establish a strong foundation for relying on Certificates of Analysis (CoAs) to maintain compliance.
Using Certificates of Analysis (CoAs) for Compliance
Once suppliers are qualified, CoAs become a vital tool for ensuring compliance. CoAs can simplify raw material testing when manufacturers set up formal programs to audit supplier-provided certificates for bulk pharmaceutical chemicals [4]. However, regular independent testing is necessary to confirm the accuracy of these CoAs.
Each CoA should be evaluated against established acceptance criteria, such as USP, NF, or ACS standards [8]. For active pharmaceutical ingredients, manufacturers should compare impurity profiles from early clinical or biobatch materials with full-scale production batches to ensure consistent quality [4]. The reliability of any CoA hinges on the integrity of the laboratory data behind it. Systems must be in place to secure electronic records, preventing unauthorized access or data deletion [7].
For example, in July 2022, the FDA issued a Warning Letter to Jost Chemical Co. for cGMP violations related to laboratory data integrity. The company lacked adequate password controls and safeguards against data deletion, leading the FDA to recommend hiring a cGMP consultant. The agency also warned that new applications listing the company as a manufacturer could face rejection until compliance issues were resolved [7].
Benefits of Working with Trusted Suppliers
Partnering with reliable suppliers simplifies compliance and reduces the need for extensive in-house testing. According to ICH Q7 guidelines, manufacturers are ultimately responsible for the quality of all raw materials and APIs, even when sourced through contract manufacturing organizations or third-party suppliers [10]. The FDA’s Q7A guidance reinforces this, stating that "Quality measures should include a system for testing raw materials, packaging materials, intermediates, and APIs" [9].
Trusted suppliers play a crucial role in managing the natural variability found in excipients and biologically derived materials, which can impact blend uniformity, stability, and manufacturability. They also ensure that any changes to raw material sources or critical attributes are handled through established change control processes, minimizing the risk of regulatory issues.
For example, Allan Chemical Corporation brings over 40 years of experience to the table, offering compendial-grade materials (USP, FCC, ACS, NF) with meticulous documentation. Their direct relationships with vetted manufacturers ensure the traceability needed for FDA audits, helping streamline qualification processes.
This content is for informational purposes only. Always consult official regulations and qualified professionals before making sourcing or formulation decisions.
Required Tests: Elemental Impurities, Assays, and Monograph-Specific Analyses
Elemental Impurity Testing
The FDA keeps a close eye on raw materials for both toxic and nutritional elements to ensure they meet regulatory standards [11].
"The FDA collects and analyzes food and other materials (foodware, vitamins, supplements, etc.) from commercial channels of trade to determine whether those materials are in compliance with applicable regulations." – FDA Elemental Analysis Manual [11]
To guide its testing, the FDA uses the Elemental Analysis Manual (EAM) for Food and Related Products, which serves as the go-to resource for analytical methods. For preparing samples, microwave digestion is employed to eliminate matrix interferences. When testing for elements like arsenic, cadmium, chromium, lead, and mercury, manufacturers should turn to EAM Section 4.7, which outlines the use of Inductively Coupled Plasma-Mass Spectrometry (ICP-MS).
| EAM Method Section | Analytical Technique | Primary Analytes |
|---|---|---|
| 4.4 | ICP-AES (Atomic Emission Spectrometry) | Various elements in food using microwave digestion |
| 4.7 | ICP-MS (Mass Spectrometry) | Arsenic, Cadmium, Chromium, Lead, Mercury |
| 4.8 | HPLC-ICP-MS | Methylmercury and Total Mercury |
| 4.10 | HPLC-ICP-MS | Arsenic species in fruit juice |
| 4.11 | HPLC-ICP-MS | Arsenic speciation in rice and rice products |
In addition to elemental impurity testing, active ingredient assays are crucial for confirming product potency.
Active Ingredient Assays
After testing for impurities, manufacturers conduct assays to measure the potency of active ingredients. These tests ensure that products meet their labeled specifications [12]. According to FDA requirements under ICH Q2(R2), assay methods must be validated for specificity (to distinguish the active ingredient from impurities), accuracy, and precision (both repeatability and intermediate precision). Assays should also be stability-indicating, meaning they can detect changes in quality over time. Typically, assay results must fall within 80%–120% of the declared content, while content uniformity tests may allow a broader range of 70%–130%. To ensure reliability, linearity is evaluated using at least five concentrations spread across the reportable range [12].
Pharmacopeial Monograph Compliance
Compliance with pharmacopeial standards is another key step in verifying product quality. Monographs from organizations like USP-NF, Ph. Eur., and ACS provide standardized methods and specifications to ensure the identity, purity, and quality of raw materials [11][13]. The FDA verifies that manufacturers adhere to these standards, which often involve validated procedures such as ICP-MS and High-Performance Liquid Chromatography. Certified Reference Materials are used for calibration and method validation, following guidelines like ICH Q3D and USP <232> [13]. Manufacturers must also check the latest revision of a monograph or method, as updates are frequent. For food-related raw materials, the FDA’s EAM offers detailed laboratory methods and practical guidance to support compliance [11].
This content is for informational purposes only. Always consult official regulations and qualified professionals before making sourcing or formulation decisions.
Recent FDA Guidance Updates and Warning Letter Case Studies
Recent FDA Regulatory Updates
The FDA has intensified its focus on raw material testing, especially for high-risk components. In May 2023, the agency mandated immediate testing for contaminants like DEG and EG in these materials. This step comes in response to global contamination incidents and has become a key priority during inspections. Manufacturers using such materials are now required to implement testing protocols without delay, as the FDA considers this critical to preventing potentially fatal contamination in drug products[1][7]. This directive builds on existing requirements by reinforcing the importance of data integrity and accountability across testing facilities.
Laboratory data integrity has also come under greater scrutiny. New guidance from the FDA stresses that computerized systems must use individualized passwords for laboratory instruments and controls, ensuring analytical data cannot be deleted or altered without authorization[7]. Furthermore, the agency now holds manufacturers fully accountable for testing conducted by third-party laboratories, treating them as extensions of the manufacturer’s own facilities. This means all third-party testing must meet CGMP standards. Robust data controls are also now a central focus, with the FDA emphasizing the need to secure analytical records against tampering.
Case Studies: FDA Warning Letter Lessons
Recent warning letters from the FDA highlight common compliance issues. One notable example is Warning Letter 320-25-42, issued to ABR Laboratory LLC in February 2025 after a September 2024 inspection. The lab was cited for failing to validate test methods as required under 21 CFR 211.165(e). Specifically, they used a plate count media that differed from the Soybean-Casein Digest Agar specified in USP <61> without demonstrating equivalence. Additionally, the lab failed to investigate a significant deviation: a refrigerator meant to store reference microorganisms (maintained at 2–8°C) reached 17.4°C for over 24 hours. As a corrective action, the FDA required a retrospective review of all drug products tested during this period of non-compliance[14].
"FDA considers contractors as extensions of the manufacturer’s own facility. Your failure to comply with CGMP may affect the quality, safety, and efficacy of the drugs you test for your clients." – Francis Godwin, Director, Office of Manufacturing Quality, Center for Drug Evaluation and Research[14]
These examples make it clear that failures in both testing procedures and data control can significantly jeopardize a manufacturer’s compliance with CGMP standards.
This content is for informational purposes only. Always consult official regulations and qualified professionals before making sourcing or formulation decisions.
Conclusion
Key Takeaways for Manufacturers and Buyers
Compliance with FDA GMP regulations for raw material testing demands strict adherence to detailed processes throughout the supply chain. Manufacturers are required to perform independent identity verification as outlined in 21 CFR 211.84, ensure complete traceability from the raw material source to the finished product, and maintain a fully independent Quality Control department separate from production activities. The FDA’s focus on data integrity means laboratory systems must have unique passwords, maintain audit trails, and prevent unauthorized data deletion – issues that have been at the center of recent enforcement actions[7].
The regulatory environment is continually shifting. For instance, as of May 2023, manufacturers must immediately test high-risk materials like Glycerin and Propylene Glycol for contaminants such as DEG and EG. This proactive stance highlights the FDA’s commitment to preventing non-compliance. Additionally, manufacturers are required to investigate all Out-of-Specification (OOS) results within 20 business days, ensuring that testing is never misused to conceal issues. These investigations must also account for any potentially affected batches[4]. The Quality Unit retains the ultimate authority to approve or reject all incoming materials, a responsibility that cannot be compromised under CGMP standards. These updates are reshaping supplier management practices to meet stricter expectations.
Beyond rigorous testing, supplier qualification is a cornerstone of compliance. Working with dependable suppliers reduces variability and mitigates risks. For example, Allan Chemical Corporation brings over 40 years of experience in supporting regulated industries. They offer compendial-grade materials (USP, FCC, ACS, NF) and maintain strong supplier relationships, supported by comprehensive documentation. Their emphasis on traceability and just-in-time delivery aligns with the stringent testing requirements manufacturers face.
Achieving GMP compliance hinges on three critical areas: thorough testing, reliable supplier partnerships, and unwavering data integrity. Manufacturers who commit to these principles safeguard product quality, ensure regulatory compliance, and strengthen their operations to handle the growing complexities of pharmaceutical production. Regular supplier audits and documentation systems, validated testing methods, are not optional – they form the backbone of a solid quality program capable of standing up to FDA scrutiny.
This content is for informational purposes only. Always consult regulatory guidelines and experts before making sourcing or formulation decisions.
FAQs
What happens if a company doesn’t follow FDA GMP requirements for raw material testing?
Non-compliance with FDA GMP standards for raw material testing can have far-reaching consequences. It puts product quality at risk, increases the chances of contamination, and can result in the production of goods that fail to meet required standards. Regulatory bodies may issue warning letters, enforce product recalls, or even shut down facilities. Additionally, companies may face legal challenges and financial penalties.
Beyond these immediate risks, failing to adhere to these requirements can harm a company’s reputation and weaken customer trust. Meeting these standards is essential – not just to protect public health, but also to shield your business from operational disruptions and regulatory setbacks.
What steps can manufacturers take to maintain data integrity in raw material testing?
Manufacturers can protect data integrity during raw material testing by enforcing strict controls and aligning with FDA guidelines. Key practices include ensuring all data remains complete, accurate, and traceable through detailed documentation and secure systems. Using controlled access measures, such as unique user logins and audit trails, helps safeguard data from unauthorized alterations.
To support reliable results, manufacturers should prioritize regular training for staff, maintain thorough process documentation, and validate all analytical methods. Cultivating a strong quality-driven work environment and adhering to current Good Manufacturing Practices (cGMP) are essential steps to ensure compliance and uphold the integrity of testing procedures.
Why are independent identity tests required even when a supplier provides a Certificate of Analysis?
Independent identity tests play a crucial role in verifying the authenticity and quality of raw materials, even when a supplier provides a Certificate of Analysis (CoA). These tests are essential for ensuring that materials comply with FDA Good Manufacturing Practice (GMP) standards and are not contaminated or substituted.
Relying only on a CoA without conducting independent verification can lead to serious risks, including regulatory non-compliance or compromised product safety. By performing these tests, companies uphold strict quality control, safeguard consumer trust, and meet regulatory requirements across industries like pharmaceuticals, food, and cosmetics.



Comments are closed