Binary solvent mixtures, like ethanol and water, are used to improve the solubility of poorly soluble drugs – an essential factor in pharmaceutical development. Predicting solubility in these systems is challenging due to complex interactions between solvents, solutes, and environmental conditions. Traditional experimental methods are resource-intensive, but computational models, particularly machine learning, offer a faster, more efficient alternative.
Key points:
- Why it matters: Poor solubility leads to ineffective drugs and higher development costs.
- Challenges: Binary mixtures involve unpredictable molecular interactions and limited datasets.
- Solutions: Machine learning models (e.g., Graph Convolutional Networks) predict solubility with high accuracy, reducing experimental requirements by up to 80%.
- Hybrid approach: Combining computational predictions with minimal experimental data optimizes resources.
- Applications: Tools like LightGBM and GCNs have been tested on drugs like aspirin and acetaminophen, achieving MAE values as low as 0.28 LogS.
These advancements are transforming drug formulation by increasing efficiency and reducing costs, while experimental validation ensures accuracy and regulatory compliance.
Data Science for Computational Drug Discovery using Python (Part 2 with PyCaret)
sbb-itb-aa4586a
Computational Methods for Solubility Prediction
Machine Learning vs Thermodynamic Methods for Solubility Prediction in Pharmaceuticals
Different computational techniques offer powerful tools to address challenges in predicting solubility for pharmaceutical formulations. These methods enhance accuracy and efficiency, making them indispensable in modern drug development.
Machine Learning Methods
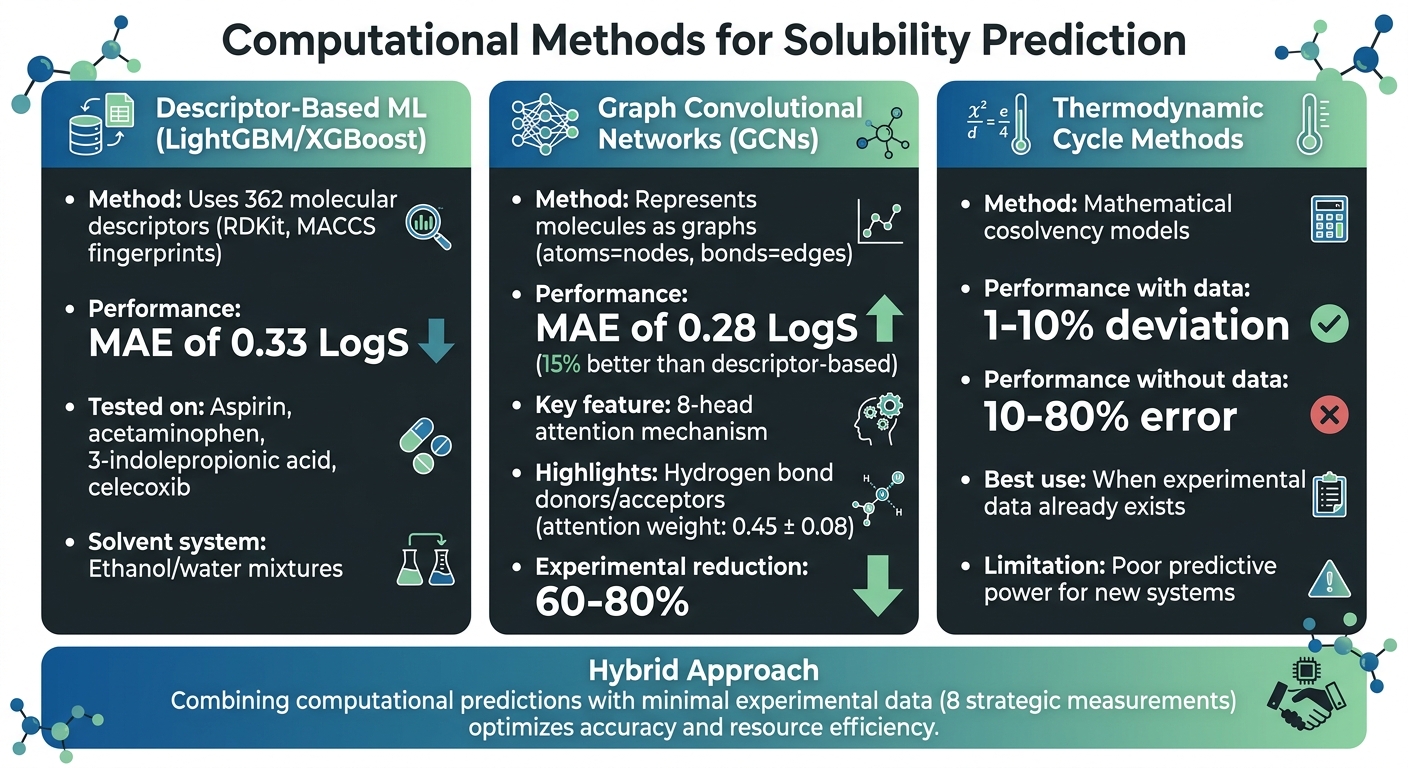
Machine learning has become a key player in predicting solubility, especially in binary systems, by analyzing experimental data. Two widely used approaches are descriptor-based models and graph-based models. Descriptor-based methods, such as LightGBM and XGBoost, rely on 362 molecular descriptors like RDKit and MACCS fingerprints to predict solubility behavior. For instance, a study published in October 2024 in the Journal of Cheminformatics reported a mean absolute error (MAE) of 0.33 LogS when testing aspirin, acetaminophen, 3-indolepropionic acid, and celecoxib in ethanol/water mixtures [2].
Graph Convolutional Networks (GCNs) take a different approach by representing molecules as graphs, where atoms are nodes and bonds are edges. This allows GCNs to directly extract structural information. A study in early 2025, published in Scientific Reports, introduced an eight-head attention GCN architecture that achieved an MAE of 0.28 LogS – 15% better than descriptor-based methods. The attention mechanism highlighted the importance of hydrogen bond donors and acceptors, assigning them the highest attention weights (0.45 ± 0.08) [1].
While machine learning provides data-driven solutions, traditional thermodynamic methods remain an alternative for solubility prediction.
Thermodynamic Cycle Methods
Thermodynamic cycle methods rely on mathematical models to link solubility with factors like temperature and solvent composition. These cosolvency models perform well when experimental data is available to fit the equations, often achieving deviations of just 1–10% [3]. However, their predictive power diminishes in the absence of prior measurements, with errors ranging from 10–80% [3].
"With regard to prediction results, the best predictions were made using the cosolvency models trained by a minimum number of experimental data points and an ab initio accurate prediction is not possible so far." – Abolghasem Jouyban, Tabriz University of Medical Sciences [3]
This limitation makes thermodynamic models less suited for entirely new systems but effective when experimental data is already available.
Combined Experimental and Computational Approaches
A hybrid approach blends the precision of experimental methods with the efficiency of computational models, making it an ideal strategy for resource optimization. In September 2025, researchers at the University of Michigan developed a hybrid workflow using LightGBM to identify organic cosolvents for benzaldehyde and limonene [4]. The process began with an aqueous solubility model (trained on AqSolDB) to filter out water-immiscible solvents based on a miscibility threshold of log(S) = 0.08. Next, an organic solubility model (trained on BigSolDB) ranked the remaining candidates [4]. This two-stage method reduced experimental work while maintaining accuracy for these specific compounds.
By combining computational predictions with targeted experimental validation, scientists can focus on the most promising solvent combinations. This is especially useful when dealing with costly or scarce drug candidates, streamlining the formulation process and conserving resources.
This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Machine Learning for Binary Solvent Mixtures
Training Data and Feature Selection
Developing machine learning models for binary solvent systems requires extensive datasets that connect molecular structures to solubility data. Typically, these models are trained using around 27,000 experimental solubility measurements, encompassing 123 solutes and 110 binary solvent mixtures. Key experimental conditions, such as temperature (ranging from 273 to 373 K) and mixing ratios, are also included in the datasets [1][2].
Two main approaches dominate training strategies. Descriptor-based models use predefined features, like RDKit descriptors and MACCS fingerprints, to capture properties such as melting temperature, LogP (a measure of lipophilicity), polarity, and hydrogen bond donor/acceptor counts [1][2]. On the other hand, Graph Convolutional Networks (GCNs) take a different path by learning directly from molecular structures. These networks treat atoms as nodes and bonds as edges, enabling them to recognize intricate structural relationships that descriptors might overlook [1].
To improve model accuracy and avoid overfitting, it’s crucial to eliminate zero-variance features or those with high correlation (r > 0.8) in high-dimensional datasets [2]. Hybrid models, which combine machine learning with thermodynamic data (like COSMO-RS predictions), have shown promising results. For instance, a hybrid Random Forest model achieved an RMSE of 0.65, outperforming a purely data-driven approach that had an RMSE of 0.75 [5].
These carefully selected features lay the groundwork for building reliable and accurate predictive models.
Performance Metrics and Validation
In pharmaceutical applications, machine learning models must undergo rigorous evaluation to ensure reliability. Metrics like Mean Absolute Error (MAE) are commonly used, alongside Root Mean Square Error (RMSE) and the Pearson correlation coefficient (r) [1][2]. Current top-performing models achieve MAE values between 0.28 and 0.33 LogS units, with GCNs outperforming descriptor-based methods by improving predictive accuracy by 15.2% [1].
To test how well these models handle new chemical environments, scaffold splits (for new molecular frameworks) and solvent splits (for new solvents) are used. Random splits often yield lower MAEs (~0.28), but solvent-based splits result in higher errors (~0.41), highlighting the difficulty of predicting solubility in unfamiliar conditions [1][5]. Some models also use Monte Carlo dropout to estimate uncertainty, which helps gauge prediction confidence [1].
Even small amounts of experimental data can significantly enhance predictions. For example, when tested on a GlaxoSmithKline solute across 16 solvents, a model’s RMSE dropped from 0.75 (with no prior data) to 0.56 after incorporating just four experimental measurements. This "drip-feeding" strategy demonstrates how adding relational data across solvents can refine model accuracy [5].
Pharmaceutical Application Examples
Real-world applications highlight the impact of these techniques on pharmaceutical formulation. In October 2024, the Journal of Cheminformatics published a study evaluating LightGBM and XGBoost models on four drug compounds: aspirin (ASA), acetaminophen (ACM), 3-indolepropionic acid (IPA), and celecoxib (CXB). These models predicted solubility in ethanol/water mixtures at 298.15 K and 313.15 K across three solvent ratios. LightGBM achieved MAEs of 0.20 for ASA and ACM, and 0.50 for IPA. However, celecoxib predictions were less accurate due to significant deviations in 8 out of 15 key features from the training data [2].
A 2025 study in Scientific Reports explored GCNs enhanced with multi-head attention mechanisms [1]. An eight-head attention GCN achieved an MAE of 0.28 LogS. The attention mechanism analysis revealed that hydrogen bond donors and acceptors were given the highest weights (0.45 ± 0.08), emphasizing their importance in solubility prediction [1].
"This research establishes GCNs as powerful tools for accelerating pharmaceutical formulation development, potentially reducing experimental requirements by 60–80% while providing interpretable molecular insights through attention mechanisms." [1]
These examples demonstrate how machine learning is transforming pharmaceutical formulation by saving time and reducing the need for extensive experimental data, all while offering detailed molecular insights.
This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Experimental Validation and Practical Considerations
Designing Experiments for Binary Solvent Systems
When it comes to validating solubility predictions, less can sometimes be more. Researchers from Tabriz University of Medical Sciences studied 56 solubility datasets (totaling 3,488 data points) and determined that 8 experimental measurements are sufficient to train predictive models effectively. They found that using these 8 data points reduced the mean percentage deviation from 17.7% to 15.5%. Adding a ninth data point showed no significant improvement (p > 0.88)[10]. This approach is particularly helpful for pharmaceutical teams dealing with limited materials or costly drug candidates.
Choosing the right experimental method depends on the solubility range of the compound:
- Shake-flask method: Best for solubility levels above 10 mg/L, following OECD 105 guidelines.
- Column elution or slow-stir methods: More accurate for compounds with solubility below 10 mg/L.
- CheqSol technique: Uses automated titration to measure both intrinsic and kinetic solubility for ionizable compounds by adjusting pH until precipitation or dissolution occurs.
These methods differentiate between thermodynamic solubility (maximum concentration at equilibrium) and kinetic solubility (concentration at precipitation onset). This distinction is critical for regulatory validation[2].
By tailoring experimental designs to specific needs, teams can better evaluate the performance of predictive models.
Comparing Experimental and Predicted Results
Experimental results often highlight where predictive models fall short. For example, a prospective study (October 2024) demonstrated that models accurately predicted the solubility of ASA and ACM (mean absolute error < 0.5 LogS). However, deviations in celecoxib predictions revealed challenges when feature sets between the model and solutes are not well-aligned[2].
"This prospective study demonstrated that the models accurately predicted the solubility of solutes in specific binary solvent mixtures under different temperatures, especially for drugs whose features closely align within the solutes in the dataset." – Journal of Cheminformatics[2]
Errors in experimental data, especially for low-solubility compounds, can reach up to 1.0 log unit[7][9]. Understanding these limitations helps teams set realistic expectations and refine their model validation strategies.
These insights highlight the importance of aligning experimental and computational efforts to meet regulatory standards.
Regulatory and Practical Requirements in Pharmaceuticals
For pharmaceutical development, experimental validation ensures that solubility predictions meet both regulatory and practical requirements. Since nearly 70% of candidate molecules face solubility challenges, this step is essential for both compliance and effective formulation[8].
Regulatory bodies require that computational predictions, particularly in binary solvent systems, be supported by reproducible experimental data. Adjusting solvent combinations and ratios in these systems provides flexibility during formulation. To enhance reliability, uncertainty quantification tools like Monte Carlo dropout can flag predictions needing further experimental verification. Meanwhile, technologies such as Graph Convolutional Networks (GCNs) may reduce experimental demands by up to 60–80%[1].
That said, for high-value compounds or limited material scenarios, a combination of minimal experimental design (8 strategic data points) and computational modeling offers a practical, cost-effective solution. This ensures compliance while optimizing resources.
This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
Conclusion
Key Takeaways
Advances in computational methods have significantly improved solubility predictions for binary solvent mixtures. Graph Convolutional Networks (GCNs) now achieve a 0.28 LogS Mean Absolute Error (MAE), reflecting a 15% improvement in accuracy, while cutting experimental requirements by 60–80% [1].
By integrating experimental data with computational predictions, hybrid approaches can further refine these models. This is particularly critical since nearly 70% of newly developed drugs face challenges related to poor aqueous solubility [6].
Future Directions in Solubility Prediction
With these validated metrics as a foundation, future research aims to enhance model interpretability and expand the scope of applications. Emerging structure-based learning techniques now incorporate features like uncertainty quantification and multi-head attention mechanisms. These advancements allow for accurate predictions across a temperature range of 273–373 K and in complex binary mixtures [1]. Importantly, these models provide insights into molecular features – such as hydrogen bond donors and acceptors – that play a pivotal role in solubility [1].
"This research establishes GCNs as powerful tools for accelerating pharmaceutical formulation development, potentially reducing experimental requirements by 60–80% while providing interpretable molecular insights through attention mechanisms." – Scientific Reports [1]
These innovations are expected to streamline drug development by reducing material costs and shortening the time to market.
Working with Trusted Chemical Suppliers
While computational tools open new possibilities for formulation development, the quality of materials used remains essential. Selecting the right solvent mixtures requires high-purity chemicals that align with computational assumptions. Allan Chemical Corporation (https://allanchems.com) offers technical-grade and compendial-grade chemicals (USP, FCC, ACS, NF) with over 40 years of experience in pharmaceutical applications. Their just-in-time delivery and strong relationships with manufacturers help researchers secure the materials needed for both experimental validation and large-scale production.
Collaborating with dependable suppliers ensures that experimental data aligns with computational models, supporting both regulatory compliance and efficient formulation processes.
This content is for informational purposes only. Consult official regulations and qualified professionals before making sourcing or formulation decisions.
FAQs
How do I choose the best ethanol/water ratio for a specific drug?
To find the best ethanol/water ratio for a drug, computational tools like the Jouyban-Acree model can be used to estimate solubility by leveraging data from pure solvents. Other methods, such as Abraham descriptors or partial solubility parameters, are also valuable for this purpose. The process typically involves analyzing available solubility data, simulating different ratios using computational models, and then conducting experimental validation. This approach helps pinpoint a ratio that optimizes solubility while ensuring stability and meeting formulation requirements.
What’s the minimum experimental data needed to trust an ML solubility model?
To rely on a machine learning (ML) solubility model, it’s crucial to start with a small but carefully constructed dataset. This dataset should closely represent the specific solvent system and temperature range you’re working with. Research indicates that even a limited amount of well-selected data can produce dependable predictions when used correctly.
How can I tell if a solubility prediction will fail on a new solvent system?
Predicting whether solubility predictions will hold up in a new solvent system involves evaluating several factors. The quality and range of the training data play a crucial role. If the model was trained on data that closely resembles the new system, predictions are more likely to be accurate. However, when the new solvent system differs significantly in chemical properties or composition from the training data, reliability can drop. This is why it’s critical to validate predictions against experimental results, especially when working with unfamiliar systems.



Comments are closed